
目前,在抗炎的临床治疗中,阿司匹林、布洛芬和萘普生等非甾体抗炎药 (NSAID) 扮演了重要角色。然而,NSAIDs是COX-1/2的非特异性抑制剂,所以在达到抗炎镇痛目的的同时,也可能会引起广泛的不良反应,比如损害胃肠道并导致肾脏、心血管和肝脏功能障碍等。此后,科学家们又开发了COX-2选择性小分子抑制剂(昔布),然而,数据显示,长期服用昔布与严重心脏副作用有关。
为了减轻非选择性COX抑制相关副作用,寻找新的靶标,开发新一代抗炎疗法成为研发重点。
1. 从COX到EP2
环氧合酶 (COX) 是花生四烯酸 (AA)合成前列腺素的关键酶。COX目前已知的异构体有两种:COX-1和COX-2,分别由ptgs1和ptgs2编码。CoX-1表达于血管、胃、肾和血小板等绝大多数组织,参与血小板聚集、血管舒缩、胃黏膜血流以及肾血流的调节,以维持细胞,组织和器官生理功能的稳定。COX-2通常在大多数正常组织和器官中以低基础水平表达。
在炎症或有害刺激下,花生四烯酸(AA,一种20碳脂肪酸)通过cPLA2从细胞膜释放,然后通过COX转化为PGH2。短寿命PGH2被组织特异性前列腺素合成酶进一步催化为五种类型的前列腺素:PGD2、PGE2、PGF2α、PGI2和TXA2。负责将PGH2 转化为PGE2的三种同工酶是mPGES-1、mPGES-2和cPGES(图1)。
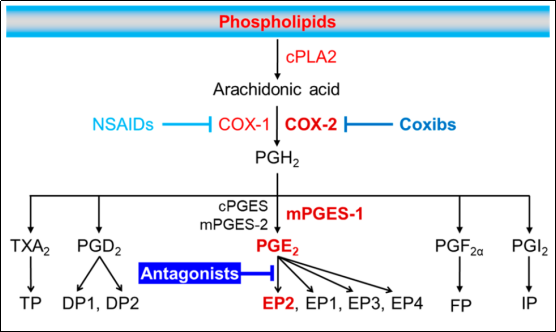
图1. COX-2/mPGES-1/PGE2/EP2信号通路(图片来源:J. Med. Chem.)
有研究表明,靶向诱导型mPGES-1可抑制COX-2衍生的PGH2合成PGE2,不会影响其他类型的前列腺素,被认为比抑制COX-2本身更具特异性。然而,由于mPGES-1酶的序列和结构的种间差异,设计和测试mPGES-1酶抑制剂非常复杂。
因此,为了避免因抑制PGE2生物合成酶而引起的并发症,下游的PGE2受体EP2被认为有望作为替代靶标,成为更具特异性的靶点。
2. PGE2/EP2 信号通路
前列腺素受体EP2可与 Gs (由α、β和γ 亚基组成的异源三聚体)组成蛋白复合物。PGE2对EP2受体的激活将Gs复合物迅速分离为Gα和Gβγ,反过来又调节了不同的下游信号分子,这些分子也可以与许多其他途径进行交互(图2)。EP2受体的激活既可以导向G-蛋白偶联信号通路,也可以导向独立信号通路,这些通路通常与其他通路(包括 EGFR介导的信号传导)进行交互。因此,可以通过靶向EP2受体开发选择性小分子拮抗剂以缓解由EP2受体介导的下游病理过程。
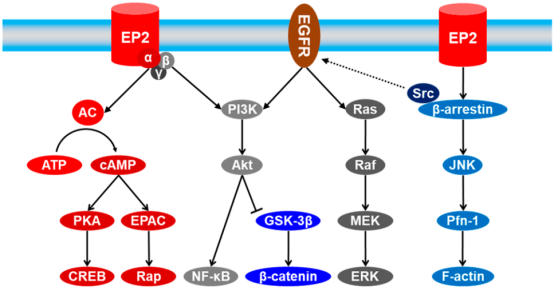
图2. EP2信号通路与其他通路的联系(图片来源:J. Med. Chem.)
3. 多家企业布局EP2拮抗剂
EP2基因消融 (EP2–/–)是早期研究中的一种抑制策略,用于阐明EP2受体的生理和病理作用。虽然非常有用,但它也因小鼠的发育和其他稳态调整而变得复杂,导致高血压和产仔数减少。
已广泛使用的EP2受体小分子激动剂包括布托前列素、CP-533536、CAY10399、ONO-AE1-259 和 C-9(图 3)。然而,布托前列素对EP2的选择性仅约为EP3的18倍;CP-533536 (evatanepag)对EP2的选择性约为EP4的64倍;CAY10399和ONO-AE1-259对EP2 具有高度选择性但具有类前列腺素样结构;C-9对其他PGE2受体的选择性相当高,但对TP受体的选择性不到四倍。
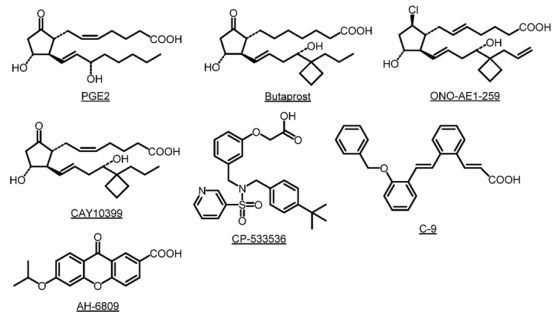
图3. 已用于研究EP2受体的常规配体和化合物(图片来源:J. Med. Chem.)
此外,还有报道具有非前列腺素结构的EP2正变构调节剂,并提供了替代化学探针来研究PGE2存在下的受体;然而,其药代动力学具有不确定性,阻碍了在体内的使用。
3.1.辉瑞开发的EP2拮抗剂
2011年,辉瑞(Pfizer)报告了其首个EP2选择性拮抗剂化合物1(图4),该拮抗剂在体外和体内试验中均显示出活性。在不存在PGE2或其他激动剂的情况下,化合物1不显示任何激动活性,也不会对EP2产生任何影响。这些结果表明它特异性地抵消了作用于EP2受体的PGE2或其他激动剂的作用。两年后,辉瑞报告了其第二个 EP2 选择性拮抗剂,化合物2。

图4. 由辉瑞开发并在动物模型中测试的选择性EP2拮抗剂(图片来源:J. Med. Chem.)
3.2.埃默里开发的化合物
2008年,埃默里大学的研究人员利用一组基于细胞的时间分辨荧光共振能量转移 (TR-FRET) 分析cAMP形成,对262371种化合物进行了高通量筛选 (HTS)。由于当时缺乏真正的选择性EP2拮抗剂,他们的目标是开发选择性抑制 EP2受体的化合物,用于研究长期癫痫引起的脑部炎症。因此,一系列小分子被鉴定为人类EP2受体的竞争性拮抗剂。其中,化合物3(图5)有效,其功能性Schild KB为2.4 nM,可拮抗PGE2,血浆半衰期(t1/2)为0.6小时,脑与血浆之比为0.3。随后的构-效关系发现了相关衍生物(4~7)(图5)。
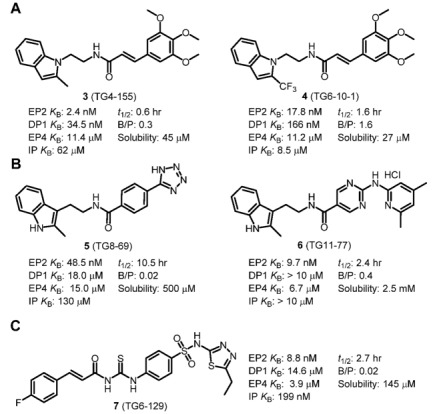
图5. 由埃默里大学开发并在临床前模型中广泛评估的EP2拮抗剂(图片来源:J. Med. Chem.)
3.3.安进开发的化合物
2015年,Amgen的研究人员通过高通量筛选鉴定出一种新型 EP2 拮抗剂,化合物8(图6)。化合物8对人类、小鼠和大鼠受体具有轻度至中度效力。在选择性测试中,其EP2/EP1受体的选择性 400,对EP2/EP3的选择性~300,但对EP2/EP4(PGE2的另一种 Gαs 偶联受体)的选择性只有50。在构-效关系研究中,又发现了化合物9。

图6. Amgen开发的EP2 拮抗剂
4. 小结
在经历了长期使用环加氧酶-2 (COX-2) 抑制剂导致的相关副作用之后,人们广泛认为,对于炎症治疗,与其简单地关闭整个COX级联反应,不如调节下游前列腺素合成酶或受体。
长期以来,人们一直将COX-2的致病作用归因于PGE2通过其Gαs偶联的EP2受体亚型发出信号。然而,真正具有选择性的EP2拮抗剂直到2011年才出现。这些小分子提供了全新的治疗或研究方法,可以帮助研究人员更好地了解炎症相关疾病中的EP2受体。它们在临床前模型中的应用也重塑了人们对PGE2/EP2信号作为健康和疾病炎症节点的认识。
今年,在选择性EP2拮抗剂发现10周年之际,探索它们作为下一代抗炎疗法候选药物的潜力才刚刚开始显现,期待未来能造福更多炎症患者。
参考文献
Sluter, Madison N., Ruida Hou, Lexiao Li. et al. EP2 Antagonists (2011–2021): A Decade’s Journey from Discovery to Therapeutics. J. Med. Chem. 2021, 64, 11816–11836