
在ADC中,单克隆抗体(mAbs)充当有效载荷(细胞毒性药物)的载体,为表达靶细胞表面抗原的细胞提供靶向作用。ADC与靶细胞表面的抗原结合后,被内化并被转运到溶酶体室降解,释放有效载荷的。
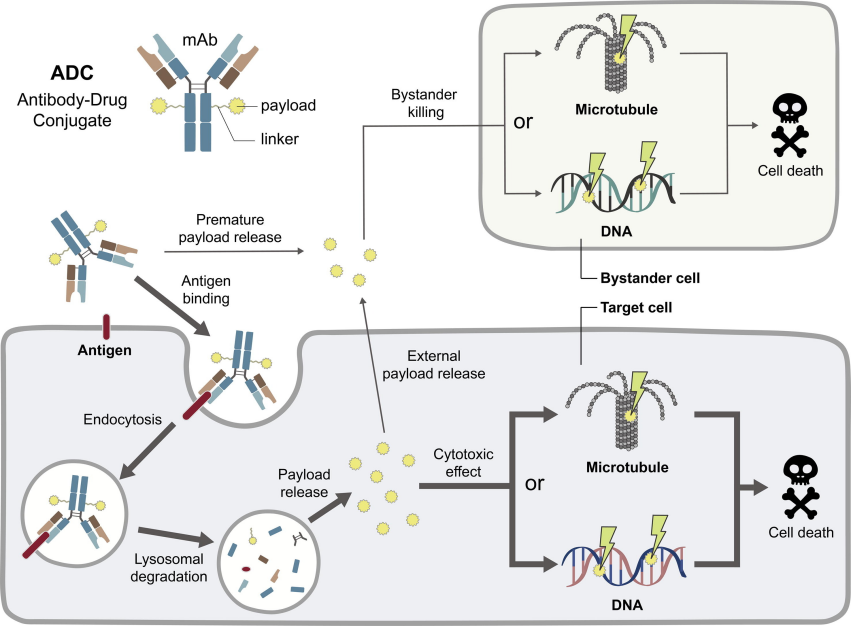
内化的程度取决于抗体对靶标的亲和力和可用于结合的抗原的密度。高度的内化并不一定有利于实体肿瘤的治疗,因为ADC的快速降解限制了其穿透肿瘤团块的能力。因此,抗体的最佳亲和力取决于靶标的性质。非内在化的adc也可以通过所谓的“旁观者效应”来显示治疗效果,即有效载荷能够渗透邻近细胞的细胞膜。
mAb关系到ADC的靶标特异性(控制细胞毒性药物向靶细胞/靶组织的递送)、靶标结合亲和力(影响实体肿瘤中的ADC分布)、免疫原性(影响治疗潜力)、大小(决定PD和PK特性)和DAR(影响治疗窗口),因此选择合适的mAb对于成功开发用于临床开发的ADC至关重要。
在ADC技术刚起步的时期,大多数药物使用的是鼠源抗体,但是鼠源抗体会导致严重的免疫原性,于是出现了嵌合抗体。嵌合单克隆抗体是通过将人类Fc区域与啮齿动物可变区域混合,具有理想的治疗结果,但是不能完全避免免疫原性的问题。因此,新一代的技术将小鼠来源的mAb的比例进一步限制在互补性决定区域(CDR)上,从而产生了所谓的“人源化单克隆抗体”。最后,应用基因工程技术进步(例如,大肠杆菌中人可变链区域的生产以及噬菌体展示技术)来实现全人抗体。
目前的ADC主要使用完整的IgG抗体,人源化抗体主要分为IgG1,IgG2,IgG3,IgG4几种,其不同区别如下。
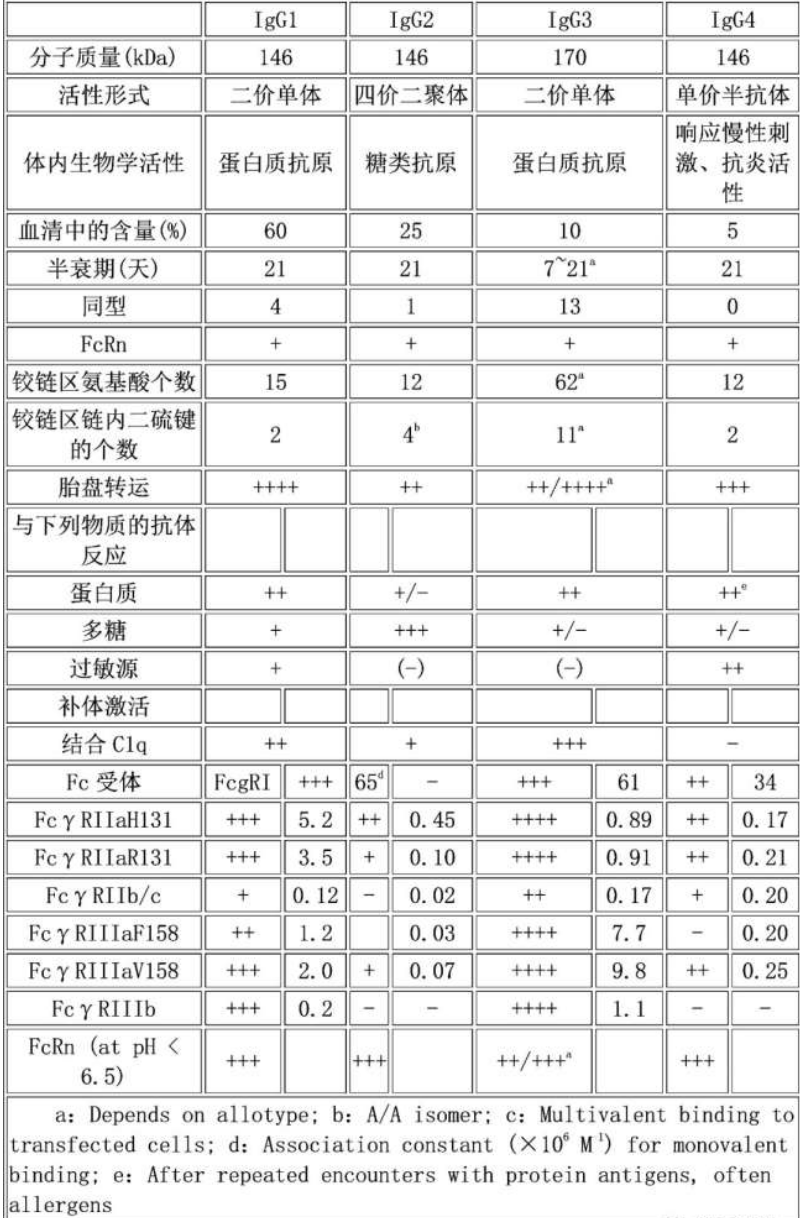
批准的 mAb 使用三种主要的同种型(IgG1、IgG2和 IgG4),每种的重链氨基酸序列不同。IgG1 同种型支持抗体依赖性细胞毒性 (ADCC) 和补体依赖性细胞毒性 (CDC),而 IgG4 启动抗体依赖性细胞吞噬作用(ADCP) 的能力有限。尽管第一个获批的 ADC Mylotarg® 使用 IgG4 mAb,但这种同种型有一些缺点,包括在体内形成包含一条重链和一条轻链的半抗体(MW ∼75 kDa) 以及 Fab 臂与内源性人的交换IgG4 抗体。IgG4 同种型用于通过在铰链区将丝氨酸突变为脯氨酸来产生gemtuzumab和 inotuzumab。
理想的ADC靶向肿瘤特异性抗原是那些在肿瘤细胞上完全和大量表达而很少在正常细胞上表达的。然而,潜在的免疫治疗靶点通常在肿瘤和正常细胞上表达,因此可用的靶标屈指可数,选择合适的肿瘤特异性抗原作为靶标仍然是ADCs临床开发的障碍,靶向表达的水平最终将决定ADC的治疗效果,如果所选抗体不完全与所选抗原结合,则与其他抗原结合可能会允许正常细胞的脱靶杀伤,并将对ADC的治疗指数产生负面影响。
在FDA目前批准使用的11种ADC中,有7种被批准用于治疗液态癌(白血病,淋巴瘤或骨髓瘤)。三种ADC被批准用于治疗乳腺癌:Enhertu®和Kadcyla®靶向人表皮生长因子受体2(HER2),而Trodelvy®靶向肿瘤相关的钙信号换能器2(TROP2)。Padcev®被批准用于晚期/转移性尿路上皮癌,靶向nectin-4。

图源Factors influencing the choice of monoclonal antibodies for antibody–drug conjugates
国内ADC药物研究的靶点也紧紧跟随FDA批准的规律,针对HER2的研究最多,接着是TROP2。
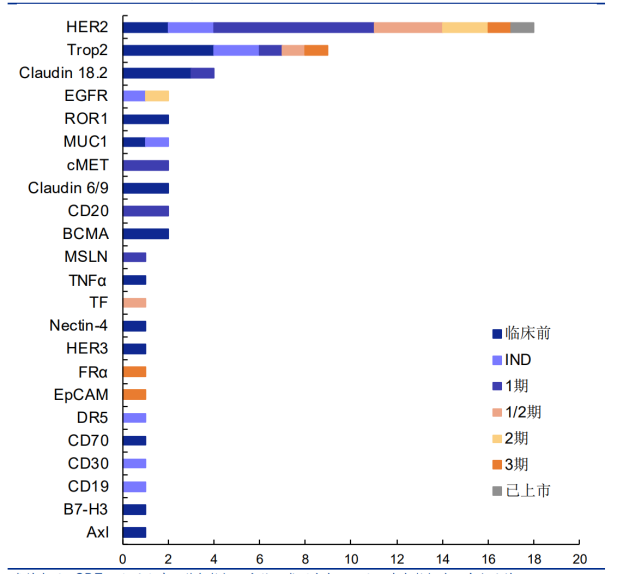
图为不完全统计,图源https://pdf.dfcfw.com/pdf/H3_AP202108041508020566_1.pdf?1628095342000.pdf
天然抗体
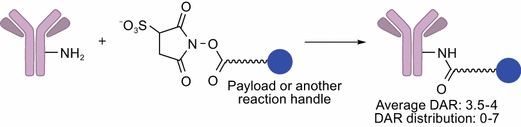
天然的抗体表面上有可以与连接子反应的氨基酸残基,比如赖氨酸残基,半胱氨酸残基。这些反应残基在一定程度上影响着ADC的DAR值。
赖氨酸共轭
IgG上大约有80个赖氨酸残基,大约10个残基可以作为接头偶联的位点。因此,这种共轭模式通常使ADC具有可变的DARs和多个缀合位点。在这种偶联模式下,一些在抗体-抗原相互作用中至关重要的赖氨酸残基可能会被修饰,导致结合亲和力降低。因此,使用这种偶联方法构建的ADC的非均相混合物可能导致治疗指数差。并且赖氨酸偶联的ADC药物一致性很差,比如在美登素型ADC的中,平均DAR为3.5-4,分布在0-7之间。高DAR可以增加效力,但也有增加聚集,清除率和循环过程中有毒有效载荷过早释放的风险,低DAR的药物又起不到相应的穿透作用。
半胱氨酸共轭
半胱氨酸共轭产生的ADC在DAR分布上比赖氨酸共轭更有优势,其可以产生较为均匀的DAR。在 IgG2 mAb的铰链区有4个二硫键,而IgG1和IgG4同种型则有2个。4个链间二硫化物通常对IgG2的结构稳定性并不重要,可以在温和的条件下选择性地还原,在保持链内二硫化物完整性的情况下得到2,4,6或8个游离硫醇。如果使用半胱氨酸残基进行连接,那么使用 IgG2 mAb 使得每个抗体能偶联更多有效荷载。
工程抗体
利用天然氨基酸残基进行偶联相对来说DAR的稳定性较低,如果想得到严格控制的DAR,那么利用基因工程在抗体中加入天然/非天然氨基酸是一个不错的选择。
引入天然氨基酸
Genentech 的科学家开发了 THIOMAB™,这是一种使用定点诱变在 mAb 中引入半胱氨酸残基的技术。该技术将 DAR 的一致性提高到 2。该技术的另一个显着贡献是它提高了使用抗体-siRNA 偶联物 (ARC) 的适用性。
引入非天然氨基酸
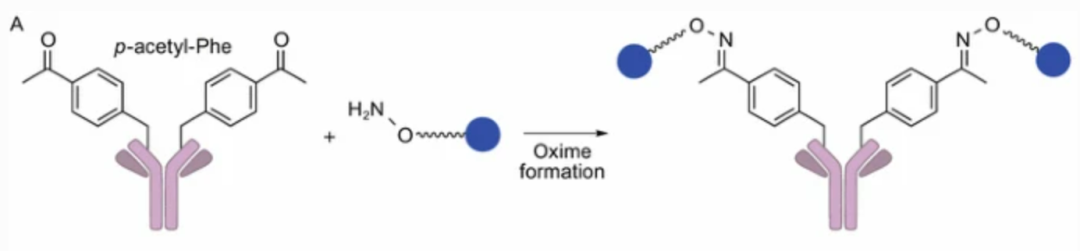
对乙酰基苯丙氨酸残基
Schultz和同事利用蛋白质表达系统(细菌,酵母和哺乳动物细胞),其中含有羰基的对乙酰基苯丙氨酸通过引入独特的密码子-tRNA合成酶进行遗传编码。使用其中任一表达系统生产含有对乙酰基苯丙氨酸残基的工程抗体,引入的羰基与烷氧胺官能化连接子反应以提供肟共轭ADC。
引入叠氮化物基团
引入对叠氮甲基-L-苯丙氨酸和N6-(2-叠氮基乙氧基)羰基)-L-赖氨酸。掺入的叠氮化物基团用于通过铜催化的Huisgen环加成(通常称为“点击化学”)与炔烃官能化连接子共轭,以提供三氮唑连接的ADC。Zimmerman等人使用这种方法将单甲基尿抑素F(MMAF)与曲妥珠单抗偶联,从而提供了有效的ADC。
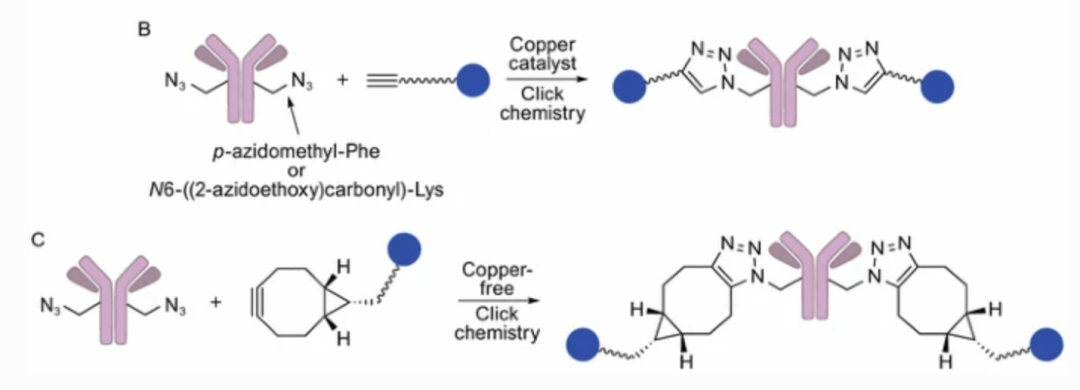
然而,基于非天然氨基酸的方法通常需要特殊的技术和生物制剂用于基因工程过程,并且掺入的非天然氨基酸残基可能会引发不必要的免疫反应,因此可能还有很长一段时间的路要走。
引入LPXTG(X:任何氨基酸)基序
自金黄色葡萄球菌的分选酶A识别LPXTG(X:任何氨基酸)基序,切割苏氨酸 - 甘氨酸(T-G)键,并附着含有寡甘氨酸(oligo-G)的分子。
Beerli及其同事将识别基序LPEG引入到各种单克隆抗体的轻链和重链的C端。然后,在分选酶A存在下,将含有五-G的小分子有效载荷单甲基尿抑素E(MMAE)偶联到mAbs上。所得偶联物(DAR:约3.2,单体含量:>96%)对抗体与对应抗原的结合没有不良影响,并且,这些偶联物发挥的体外细胞杀伤活性与传统ADC偶联方法产生的相应偶联物相当。
[1] Factors influencing the choice of monoclonal antibodies for antibody–drug conjugates